abyssMG
M1: MAG reconstruction
Blandine Trouche October 2023
- 1- Quality filtering
- 2- Running Simka and defining co-assembly groups
- 3- Co-assembling with Megahit and binning with Concoct
- 4- Refining bins
- 5- Dereplicating bins
- 6- Creating the databases with the dereplicated AOA MAGs
- 8- Create single MAGs profiles for vizualisation and further analysis
- 9- Run variability profiling for all MAGs
1- Quality filtering
conda activate anvio-7.1
# run the workflow until gzip of quality filtered fastqs
# config can be find in the archive available on github
anvi-run-workflow -w metagenomics \
-c 1_config_qc.json \
--additional-params \
--until gzip_fastqs \
--cores 12 \
--jobs 6 \
--keep-going --rerun-incomplete \
--unlock
conda deactivate
2- Running Simka and defining co-assembly groups
# Generating the Simka input
cd 01_QC
for R1 in *-QUALITY_PASSED_R1.fastq.gz
do
NAME=$R1
NAME="${NAME/-QUALITY_PASSED_R1.fastq.gz/}"
NAME="${NAME/..\/01_QC\//}"
R2=$R1
R2="${R2/R1.fastq.gz/R2.fastq.gz}"
echo -e "$NAME":" ""$R1"";""$R2" >> simka_input.txt
done
# Running Simka
conda activate simka
cd 01_QC/
simka -nb-cores 36 -max-memory 500000 \
-in simka_input.txt -out ../10_SIMKA/ -out-tmp $SCRATCH
# Plotting Simka's output
library(vegan)
## Loading required package: permute
## Loading required package: lattice
## This is vegan 2.6-4
library(ggplot2)
library(ggdendro)
library(dendextend)
## Registered S3 method overwritten by 'dendextend':
## method from
## rev.hclust vegan
##
## ---------------------
## Welcome to dendextend version 1.16.0
## Type citation('dendextend') for how to cite the package.
##
## Type browseVignettes(package = 'dendextend') for the package vignette.
## The github page is: https://github.com/talgalili/dendextend/
##
## Suggestions and bug-reports can be submitted at: https://github.com/talgalili/dendextend/issues
## You may ask questions at stackoverflow, use the r and dendextend tags:
## https://stackoverflow.com/questions/tagged/dendextend
##
## To suppress this message use: suppressPackageStartupMessages(library(dendextend))
## ---------------------
##
## Attaching package: 'dendextend'
## The following object is masked from 'package:ggdendro':
##
## theme_dendro
## The following object is masked from 'package:permute':
##
## shuffle
## The following object is masked from 'package:stats':
##
## cutree
library(here)
## here() starts at /Users/btrouche/Desktop/W_LM2E/SDU/abyssMG
# read metadata
metadata <- read.csv(here("10_SIMKA/simka_metadata.csv"), sep = ";", header = T, row.names = 1)
# function to format distance
format_distance <- function(distanceMatrix, numberAxis) {
distances <- as.dist(distanceMatrix)
pcoa <- cmdscale(distances, k = numberAxis, eig = TRUE)
plotData <- data.frame(pcoa$points)
relEig <- (100 * pcoa$eig / sum(abs(pcoa$eig)) )[1:numberAxis]
colnames(plotData) <- paste0("Dim", seq(1, numberAxis))
return(list(data = plotData, eig = relEig))
}
# load data
bray_Atacama <- as.matrix(read.table(file=here("10_SIMKA/mat_abundance_braycurtis.csv"), sep=";", header=TRUE, row.names=1))
### INFO HABITAT
variables = metadata$HABITAT
dataset_ids = rownames(metadata)
metadata_index = list()
for(i in 1:length(dataset_ids)){
dataset_id = dataset_ids[i]
metadata_index[[dataset_id]] = variables[[i]]
}
colors = c()
dataset_ids = rownames(bray_Atacama)
for(i in 1:dim(bray_Atacama)[1]){
dataset_id = dataset_ids[i]
colors = c(as.character(colors), as.character(metadata_index[[dataset_id]]))
}
colors_habitat <- colors
colors_habitat <- sub("\\<abyssal\\>", "royalblue", colors_habitat)
colors_habitat <- sub("\\<hadal\\>", "gold", colors_habitat)
order_habitat <- c("royalblue", "gold")
### INFO SITE
variables = metadata$SITE
dataset_ids = rownames(metadata)
metadata_index = list()
for(i in 1:length(dataset_ids)){
dataset_id = dataset_ids[i]
metadata_index[[dataset_id]] = variables[[i]]
}
colors = c()
dataset_ids = rownames(bray_Atacama)
for(i in 1:dim(bray_Atacama)[1]){
dataset_id = dataset_ids[i]
colors = c(as.character(colors), as.character(metadata_index[[dataset_id]]))
}
colors_site <- colors
colors_site <- sub("\\<Ata3\\>", "yellow", colors_site)
colors_site <- sub("\\<Ata7\\>", "green", colors_site)
colors_site <- sub("\\<Ata9\\>", "red", colors_site)
colors_site <- sub("\\<Ata10\\>", "royalblue", colors_site)
colors_site <- sub("\\<Ker_ST6\\>", "#D14285", colors_site)
colors_site <- sub("\\<Ker_ST7\\>", "#6DDE88", colors_site)
order_site <- c("yellow", "green", "red", "royalblue", "purple", "mediumorchid1", "darkgreen", "pink", "#C84248", "#38333E", "cyan2", "#D7C1B1", "orange")
### INFO HORIZON
variables = metadata$HORIZON
dataset_ids = rownames(metadata)
metadata_index = list()
for(i in 1:length(dataset_ids)){
dataset_id = dataset_ids[i]
metadata_index[[dataset_id]] = variables[[i]]
}
colors = c()
dataset_ids = rownames(bray_Atacama)
for(i in 1:dim(bray_Atacama)[1]){
dataset_id = dataset_ids[i]
colors = c(as.character(colors), as.character(metadata_index[[dataset_id]]))
}
colors_horizon <- colors
colors_horizon <- sub("\\<0_1\\>", "cyan", colors_horizon)
colors_horizon <- sub("\\<1_3\\>", "gold", colors_horizon)
colors_horizon <- sub("\\<3_5\\>", "chartreuse", colors_horizon)
colors_horizon <- sub("\\<5_10\\>", "royalblue", colors_horizon)
colors_horizon <- sub("\\<10_15\\>", "purple", colors_horizon)
colors_horizon <- sub("\\<15_30\\>", "red", colors_horizon)
order_horizon <- c("cyan", "gold", "chartreuse", "royalblue", "purple", "red", "orange")
# PCoA avec code Simka
pca_axis1 <- 1
pca_axis2 <- 2
distData <- format_distance(bray_Atacama, max(pca_axis1, pca_axis2))
x = distData$data[,paste0("Dim", pca_axis1)]
y = distData$data[,paste0("Dim", pca_axis2)]
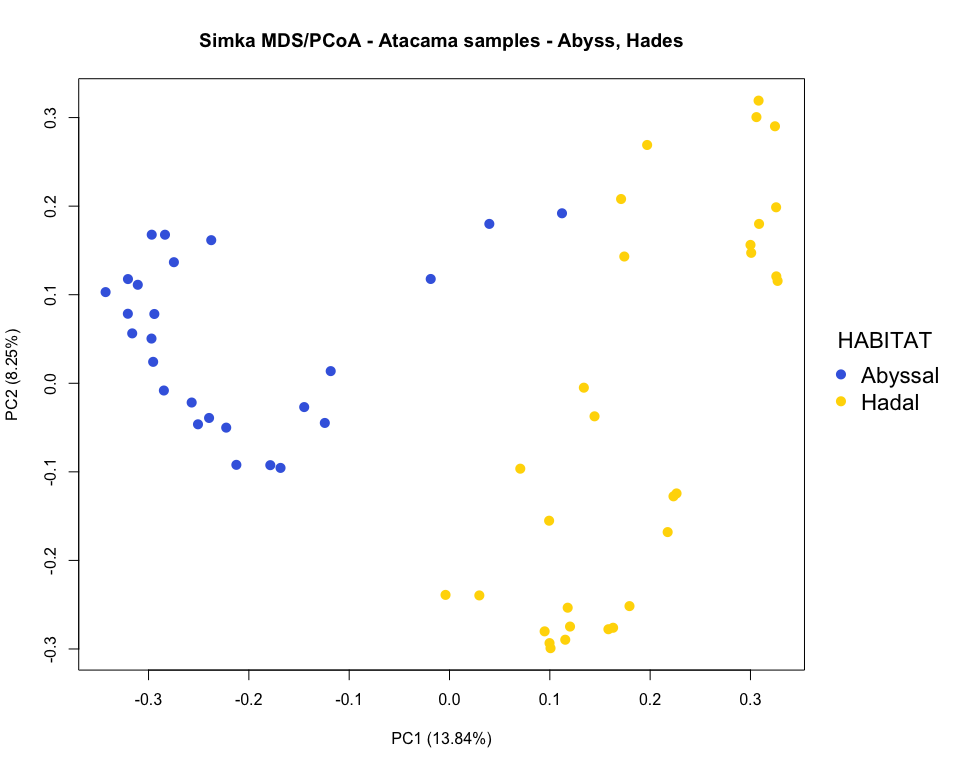
### pcoa plot colored by habitat
par(mar=c(5.1, 4.1, 4.1, 8.1), xpd=TRUE)
plot(x, y, type='n',
xlab=paste0("PC", pca_axis1, " (", round(distData$eig[pca_axis1], 2), "%)"),
ylab=paste0("PC", pca_axis2, " (", round(distData$eig[pca_axis2], 2), "%)"))
points(x, y, pch=16, cex=1.4, col=colors_habitat)
legend("right", title="HABITAT", legend = c("Abyssal", "Hadal"),
col=order_habitat, pch=16, cex=1.4, bty = "n", inset=c(-0.2, 0))
title("Simka MDS/PCoA - Atacama samples - Abyss, Hades")
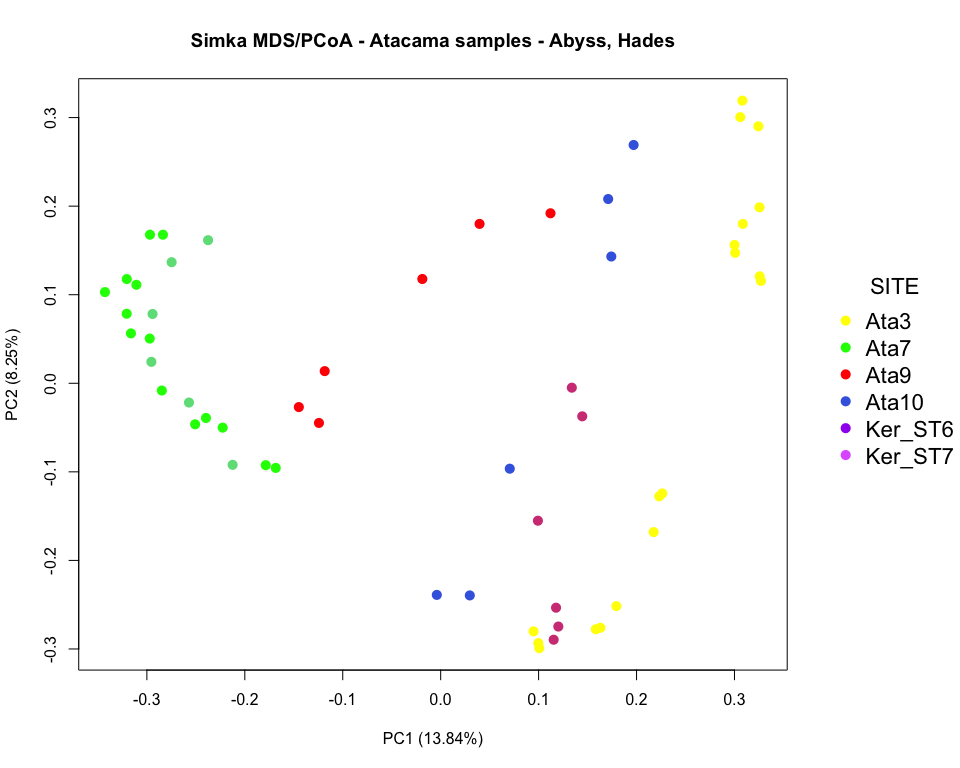
### pcoa plot colored by site
par(mar=c(5.1, 4.1, 4.1, 9), xpd=TRUE)
plot(x, y, type='n',
xlab=paste0("PC", pca_axis1, " (", round(distData$eig[pca_axis1], 2), "%)"),
ylab=paste0("PC", pca_axis2, " (", round(distData$eig[pca_axis2], 2), "%)"))
points(x, y, pch=16, cex=1.4, col=colors_site)
legend("right", title="SITE", legend = c("Ata3", "Ata7", "Ata9", "Ata10",
"Ker_ST6", "Ker_ST7"),
col=order_site, pch=16, cex=1.4, bty = "n", inset=c(-0.25, 0))
title("Simka MDS/PCoA - Atacama samples - Abyss, Hades")
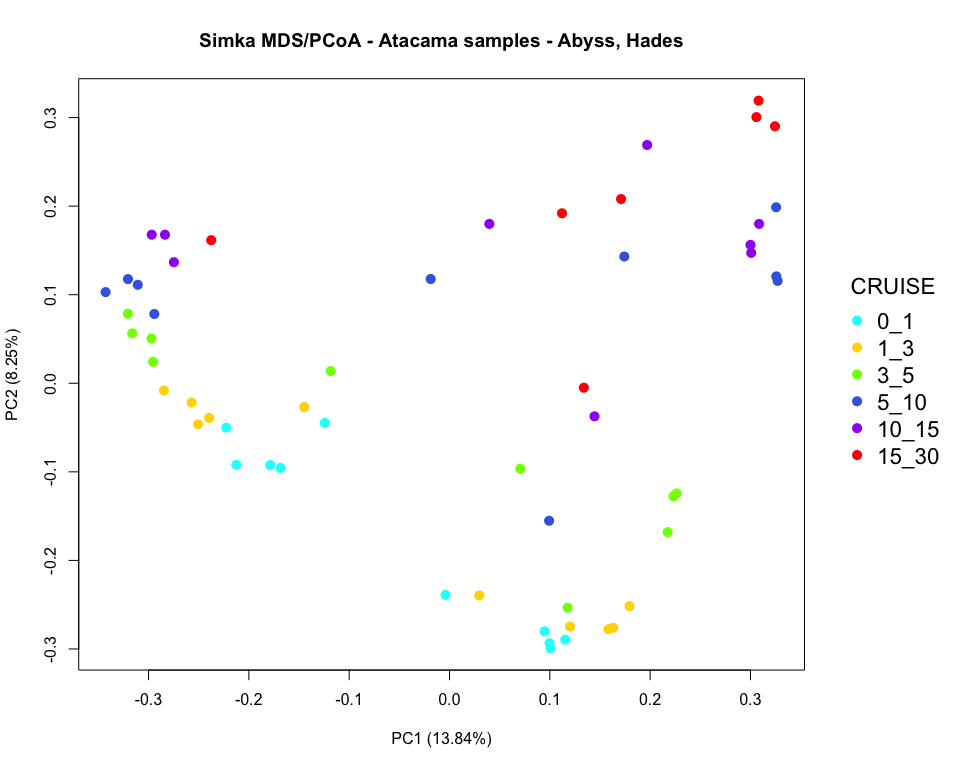
### pcoa plot colored by horizon
par(mar=c(5.1, 4.1, 4.1, 8.1), xpd=TRUE)
plot(x, y, type='n',
xlab=paste0("PC", pca_axis1, " (", round(distData$eig[pca_axis1], 2), "%)"),
ylab=paste0("PC", pca_axis2, " (", round(distData$eig[pca_axis2], 2), "%)"))
points(x, y, pch=16, cex=1.4, col=colors_horizon)
legend("right", title="CRUISE", legend = c("0_1", "1_3", "3_5", "5_10", "10_15", "15_30"),
col=order_horizon, pch=16, cex=1.4, bty = "n", inset=c(-0.2, 0))
title("Simka MDS/PCoA - Atacama samples - Abyss, Hades")
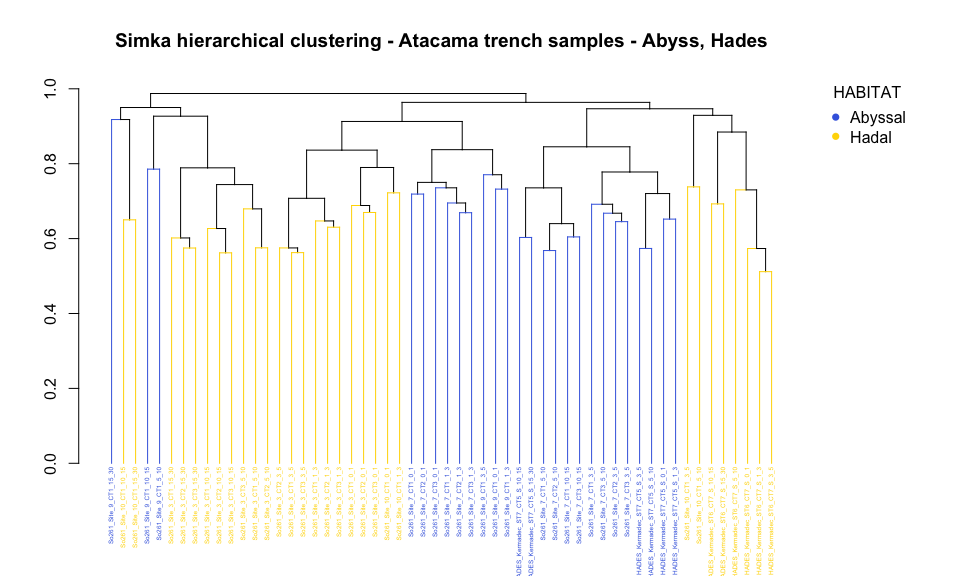
Commet_distance = as.dist(bray_Atacama)
hc <- hclust(Commet_distance)
### hclust plot colored by habitat
# Build dendrogram object from hclust results
dendo_cr3 = as.dendrogram(hc)
# set labels, labels color and size
colors_habitat = colors_habitat[hc$order]
labels_colors(dendo_cr3) <- colors_habitat
labels_cex(dendo_cr3) <- 0.4
par(mar=c(5.1, 4.1, 4.1, 8.1), xpd=TRUE)
dendo_cr3 %>% set("branches_k_color", colors_habitat) %>% plot(main="Simka hierarchical clustering - Atacama trench samples - Abyss, Hades", xlab="", sub="")
legend("topright", title="HABITAT", legend = c("Abyssal", "Hadal"), col=order_habitat, pch=16, bty = "n", inset=c(-0.15, 0))
### hclust plot colored by site
# Build dendrogram object from hclust results
dendo_cr3 = as.dendrogram(hc)
# set labels, labels color and size
#colors_site <- colors_site[order.dendrogram(dend)]
colors_site = colors_site[hc$order]
labels_colors(dendo_cr3) <- colors_site
labels_cex(dendo_cr3) <- 0.4
par(mar=c(5.1, 4.1, 4.1, 8.1), xpd=TRUE)
dendo_cr3 %>% set("branches_k_color", colors_site) %>%
plot(main="Simka hierarchical clustering - Atacama trench samples - Abyss, Hades",
xlab="", sub="")
legend("topright", title="SITE", legend = c("Ata3", "Ata7", "Ata9", "Ata10",
"Ker_ST6", "Ker_ST7"),
col=order_site, pch=16, bty = "n", inset=c(-0.2, 0))
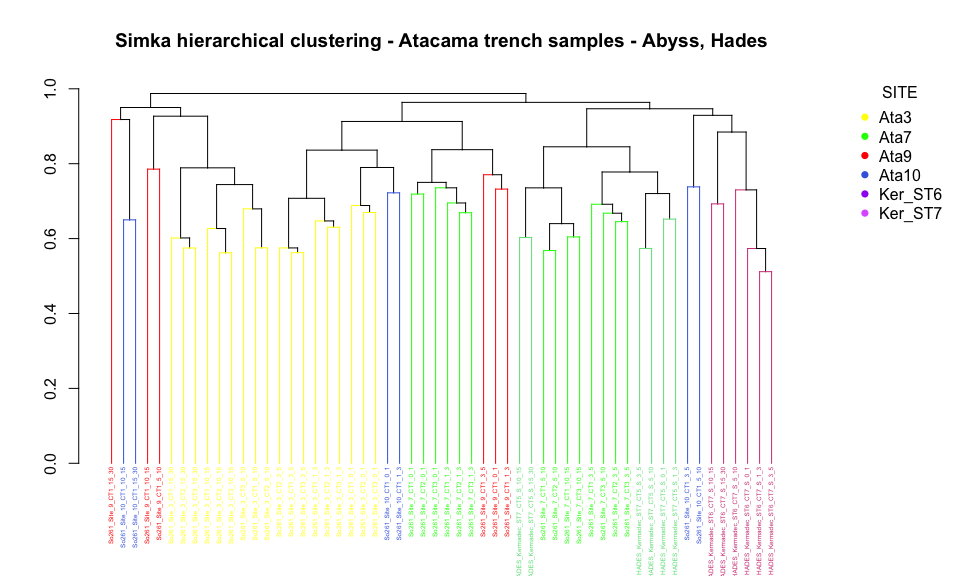
### hclust plot colored by horizon
# Build dendrogram object from hclust results
dendo_cr3 = as.dendrogram(hc)
# set labels, labels color and size
colors_horizon = colors_horizon[hc$order]
labels_colors(dendo_cr3) <- colors_horizon
labels_cex(dendo_cr3) <- 0.4
par(mar=c(5.1, 4.1, 4.1, 8.1), xpd=TRUE)
dendo_cr3 %>%
set("branches_k_color", colors_horizon) %>%
plot(main="Simka hierarchical clustering - Atacama trench samples - Abyss, Hades", xlab="", sub="")
legend("topright", title="HORIZON",
legend = c("0_1", "1_3", "3_5", "5_10", "10_15", "15_30", "30_45"),
col=order_horizon, pch=16, bty = "n", inset=c(-0.15, 0))
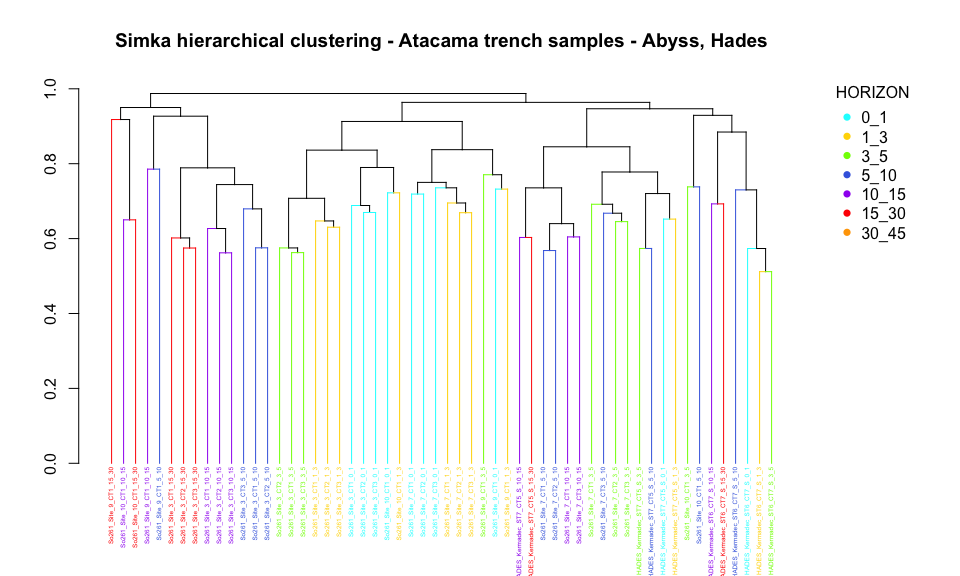
3- Co-assembling with Megahit and binning with Concoct
# running the workflow for assembly, contigs db creation, mapping, profile db creation
# remember to deactivate qc since it has been done separately
conda activate anvio-7.1
anvi-run-workflow -w metagenomics \
-c 2_config_assembly_mapping.json \
--additional-params \
--jobs 12 \
--keep-going --rerun-incomplete
# get the contig stats information
anvi-display-contigs-stats 03_CONTIGS/A7D-contigs.db 03_CONTIGS/A7S-contigs.db \
03_CONTIGS/A9S-contigs.db 03_CONTIGS/AK7-contigs.db 03_CONTIGS/H3D-contigs.db \
03_CONTIGS/H3T-contigs.db 03_CONTIGS/HAK-contigs.db 03_CONTIGS/HAS-contigs.db \
03_CONTIGS/HKT-contigs.db 03_CONTIGS/MD-contigs.db --report-as-text \
-o 03_CONTIGS/CONTIGS-STATS
# running concoct for automatic binning, limiting it to half the number of predicted bacterial bins
cat 03_CONTIGS/CONTIGS-STATS-T | while read sample
do
# determine metagenomic set names and their genomes number:
NAME=`echo $sample | awk '{print $1}'`
if [ "$NAME" == "contigs_db" ]; then continue; fi
BAC=`echo $sample | awk '{print $22}'`
NB=`expr $BAC / 2`
echo "Working on contigs db $NAME"
# do concoct:
anvi-cluster-contigs -c 03_CONTIGS/$NAME-contigs.db \
-p 06_MERGED/$NAME/PROFILE.db \
--num-threads 10 \
--driver CONCOCT \
--collection-name CONCOCT \
--clusters $NB >> 00_LOGS/$NAME-anvi-cluster-with-concoct.log 2>&1
done
4- Refining bins
Ressources from Anvi’o’s blog relating to the manual refining process
See: http://merenlab.org/2015/05/11/anvi-refine/
http://merenlab.org/2017/01/03/loki-the-link-archaea-eukaryota/
http://merenlab.org/2017/05/11/anvi-refine-by-veronika/
http://merenlab.org/data/tara-oceans-mags/
MAG quality criteria:
- finished MAG: single continuous sequence without gaps or ambiguities with consensus error rate Q50 or better
- high-quality draft MAG: >90% completion, <5% redundancy, presence of 23S, 16S, 5S rRNA genes and 18 tRNAs at least
- medium-quality draft MAG: >= 50% completion, <10% redundancy
- low-quality draft MAG: <50% completion, <10% redundancy
Estimate taxonomy based on SCGs
conda activate anvio-7.1
for GROUP in A7D A7S A9S AK7 H3D H3T HAK HAS HKT MD
do
anvi-run-scg-taxonomy -c 03_CONTIGS/$GROUP-contigs.db \
-T 5 -P 4 >> 00_LOGS/$GROUP-anvi_run_scg_taxonomy.log 2>&1
anvi-estimate-genome-taxonomy -c 03_CONTIGS/$GROUP-contigs.db \
-p 06_MERGED/$GROUP/PROFILE.db \
--metagenome-mode \
--compute-scg-coverages \
--update-profile-db-with-taxonomy >> 00_LOGS/$GROUP-anvi_estimate_genome_taxonomy.log 2>&1
done
Example of refining command lines
conda activate anvio-7.1
# first step
anvi-summarize -p 06_MERGED/A7D/PROFILE.db -c 03_CONTIGS/A7D-contigs.db -C CONCOCT -o A7D_first_summary
# choose the archaeal bins to be refined and keep going
# don't forget to ssh interactively
anvi-refine -p 06_MERGED/A7D/PROFILE.db -c 03_CONTIGS/A7D-contigs.db \
-C CONCOCT -b Bin_124 --server-only -P 8080
Once refining (focused on Archaea in our case) is done, we will rename
bins by co-assembly group, so we can regroup the bins of interest
obtained from all co-assembly groups.
We do not want to filter bins based on redundancy and completion, so no
bins will be called MAGs.
Format of bin names: “Group_Bin_number”
Renaming refined bins and summarize
for GROUP in A7D A7S A9S AK7 H3D H3T HAK HAS HKT MD
do
anvi-rename-bins -c 03_CONTIGS/$GROUP-contigs.db -p 06_MERGED/$GROUP/PROFILE.db \
--collection-to-read CONCOCT --collection-to-write ARCHAEA_REFINED \
--prefix $GROUP --report-file 06_MERGED/$GROUP/Renaming_bins_file_path \
>> 00_LOGS/$GROUP-anvi_rename_bins.log 2>&1
done
Summary will be computed for the renamed bins so we can extract the ones we want
for GROUP in A7D A7S A9S AK7 H3D H3T HAK HAS HKT MD
do
anvi-summarize -c 03_CONTIGS/$GROUP-contigs.db \
-p 06_MERGED/$GROUP/PROFILE.db \
-C ARCHAEA_REFINED \
-o 07_SUMMARY/$GROUP-renamed >> 00_LOGS/$GROUP-anvi_summarize.log 2>&1
done
Since there is no cut-off on completion or redundance for the bins we want to keep, the text file mentioning them is created by hand. This can be replicated by simply using the first column of table S4. The fasta files generated during summary (step before) are copied into REDUNDANT_BINS folder, while renaming contigs with a unique name for each of the format “Bin-name_contig-number”
mkdir -p REDUNDANT_BINS
mkdir -p REDUNDANT_BINS/02_FASTA
mkdir -p REDUNDANT_BINS/03_CONTIGS
mkdir -p REDUNDANT_BINS/04_MAPPING
mkdir -p REDUNDANT_BINS/05_ANVIO_PROFILE
mkdir -p REDUNDANT_BINS/07_SUMMARY
# go through each MAG, in each SUMMARY directory, and store a copy of the FASTA file
# with proper deflines in the REDUNDANT_BIN directory:
for BIN in `cat Archaeal_bins.txt`
do
GROUP=$(awk -F_ '{print $1}' <<< $BIN)
echo $GROUP
anvi-script-reformat-fasta 07_SUMMARY/$GROUP-renamed/bin_by_bin/$BIN/$BIN-contigs.fa \
--simplify-names --prefix $BIN \
-o REDUNDANT_BINS/02_FASTA/$BIN.fa >> 00_LOGS/$GROUP-reformat_fasta_archaea.log
done
Estimate taxonomy for each archaeal bin using CheckM
conda activate checkm
cd REDUNDANT_BINS
checkm tree 02_FASTA -x .fa -t 30 `pwd`/REDUNDANT-BINS-CHECKM-TREE
checkm tree_qa `pwd`/REDUNDANT-BINS-CHECKM-TREE -f REDUNDANT-BINS-CHECKM.txt
Create the databases for the redundant bins
conda activate anvio-7.1
cd REDUNDANT_BINS
# concatenate all contigs from the bins in one fasta file
cat 02_FASTA/*.fa > REDUNDANT-BINS.fa
# generate a contig database
anvi-gen-contigs-database -f REDUNDANT-BINS.fa
-o 03_CONTIGS/REDUNDANT-BINS-contigs.db
anvi-run-hmms -c 03_CONTIGS/REDUNDANT-BINS-contigs.db --num-threads 20
# We now want to map reads from all the metagenomes to this database
# Building the Botwie2 database
bowtie2-build REDUNDANT-BINS.fa 04_MAPPING/REDUNDANT-BINS
# going through each metagenomic sample, and mapping short reads against the 47 redundant archaeal bins
for sample in `awk '{print $1}' ../samples.txt`
do
if [ "$sample" == "sample" ]; then continue; fi
bowtie2 --threads 20 \
-x 04_MAPPING/REDUNDANT-BINS \
-1 ../01_QC/$sample-QUALITY_PASSED_R1.fastq.gz \
-2 ../01_QC/$sample-QUALITY_PASSED_R2.fastq.gz \
--no-unal \
-S 04_MAPPING/$sample-in-RBINS.sam
# convert the resulting SAM file to a BAM file:
samtools view -F 4 -bS 04_MAPPING/$sample-in-RBINS.sam > 04_MAPPING/$sample-in-RBINS-RAW.bam
# sort and index the BAM file:
samtools sort 04_MAPPING/$sample-in-RBINS-RAW.bam -o 04_MAPPING/$sample-in-RBINS.bam
samtools index 04_MAPPING/$sample-in-RBINS.bam
# remove temporary files:
rm 04_MAPPING/$sample-in-RBINS.sam 04_MAPPING/$sample-in-RBINS-RAW.bam
done
# profile each BAM file
for sample in `awk '{print $1}' ../samples.txt`
do
if [ "$sample" == "sample" ]; then continue; fi
anvi-profile -c 03_CONTIGS/REDUNDANT-BINS-contigs.db \
-i 04_MAPPING/$sample-in-RBINS.bam \
--skip-SNV-profiling \
--num-threads 16 \
-o 05_ANVIO_PROFILE/$sample-in-RBINS
done
# merge resulting profiles into a single anvi'o merged profile
anvi-merge 05_ANVIO_PROFILE/*-in-RBINS/PROFILE.db \
-c 03_CONTIGS/REDUNDANT-BINS-contigs.db \
-o 06_REDUNDANT_BINS_MERGED
# Automatically create the collection file
for split_name in `sqlite3 03_CONTIGS/REDUNDANT-BINS-contigs.db 'select split from splits_basic_info'`
do
# in this loop $split_name goes through names like this: A7D_Bin_00001_000000000001_split_00001 so we can extract the Bin name it belongs to:
BIN=`echo $split_name | awk 'BEGIN{FS="_"}{print $1"_"$2"_"$3}'`
# print it out with a TAB character
echo -e "$split_name\t$BIN"
done > REDUNDANT-BINS-COLLECTION.txt
anvi-import-collection REDUNDANT-BINS-COLLECTION.txt \
-c 03_CONTIGS/REDUNDANT-BINS-contigs.db \
-p 06_REDUNDANT_BINS_MERGED/PROFILE.db \
-C REDUNDANT_BINS
anvi-summarize -c 03_CONTIGS/REDUNDANT-BINS-contigs.db \
-p 06_REDUNDANT_BINS_MERGED/PROFILE.db \
-C REDUNDANT_BINS \
-o 07_REDUNDANT_BINS_SUMMARY
# Visualize the full redundant bin collection
ssh -L 8080:localhost:8080 trouche@penduick
anvi-interactive -p 06_REDUNDANT_BINS_MERGED/PROFILE.db \
-c 03_CONTIGS/REDUNDANT-BINS-contigs.db \
-C REDUNDANT_BINS --server-only -P 8080
# estimate taxonomy with SCGs (based on GTDB)
anvi-run-scg-taxonomy -c 03_CONTIGS/REDUNDANT-BINS-contigs.db -T 5 -P 6
anvi-estimate-scg-taxonomy -c 03_CONTIGS/REDUNDANT-BINS-contigs.db \
-p 06_REDUNDANT_BINS_MERGED/PROFILE.db \
-C REDUNDANT_BINS \
--output-file Redundant_bins_scg_taxonomy.txt
5- Dereplicating bins
# this program uses pyANI computes average nucleotide identity between MAGs by aligning them first.
# --min-alignment-fraction : this parameter removes hits between genomes if
# the alignment is weak, aka alignment fraction is under this value
# --min-full-percent-identity : this parameter removes hits between genomes if
# the full percent identity (identity*coverage) is below this value
# --similarity-threshold : this parameter determines MAGs that will be aggregated
# The input_derep.txt file was created by hand, it gives information about the location
# of files for each bin as follows (separated by tabulations):
# name bin_id collection_id profile_db_path contigs_db_path
# A7D_Bin_00002 A7D_Bin_00002 REDUNDANT_BINS 06_REDUNDANT_BINS_MERGED/PROFILE.db 03_CONTIGS/REDUNDANT-BINS-contigs.db
anvi-dereplicate-genomes --internal-genomes input_derep.txt \
--output-dir 08_DEREP_BINS --program pyANI \
--min-alignment-fraction 0.50 \
--similarity-threshold 0.95 --num-threads 16 \
--log-file 00_LOGS/anvi-dereplicate-genomes_50_95.log
6- Creating the databases with the dereplicated AOA MAGs
During this step, we will profile the nucleotide, codon and amino acid variability.
conda activate anvio-7.1
cd $HOME
mkdir NON_REDUNDANT_BINS
# create a single FASTA file for NON-REDUNDANT-MAGs
cat REDUNDANT_BINS/08_DEREP_BINS/GENOMES/*.fa > NON_REDUNDANT_BINS/02_FASTA/NON-REDUNDANT-MAGs.fa
# The next steps will be done with the snakemake workflow in references mode.
anvi-run-workflow -w metagenomics \
-c config_reference_non_redundant.json \
--additional-params \
--directory NON_REDUNDANT_BINS \
--keep-going --rerun-incomplete \
--cores 20 |& tee -a logs/workflow_log_ref.txt
### REMEMBER TO DISABLE FASTA REFORMATTING TO KEEP THE USEFUL NAMES OF CONTIGS WITH BIN INFO ###
cd NON_REDUNDANT_BINS
# create an anvi'o collection file for non-redundant MAGs:
for split_name in `sqlite3 03_CONTIGS/NR_MAGs-contigs.db 'select split from splits_basic_info'`
do
# in this loop $split_name goes through names like this:
# A7D_Bin_00001_000000000001_split_00001 so we can extract the Bin name it belongs to:
MAG=`echo $split_name | awk 'BEGIN{FS="_"}{print $1"_"$2"_"$3}'`
# print out the collections line
echo -e "$split_name\t$MAG"
done > NR_MAGs_COLLECTION.txt
# import the collection into the anvi'o merged profile database for non-redundant MAGs:
anvi-import-collection NR_MAGs_COLLECTION.txt \
-c 03_CONTIGS/NR_MAGs-contigs.db \
-p 06_NR-MAGs-MERGED/NR_MAGs/PROFILE.db \
-C NR_MAGs
anvi-estimate-scg-taxonomy -c 03_CONTIGS/NR_MAGs-contigs.db \
-p 06_NR-MAGs-MERGED/NR_MAGs/PROFILE.db \
-C NR_MAGs \
--output-file NR_archaeal_bins_scg_taxonomy.txt
# summarize the non-redundant MAGs collection:
anvi-summarize -c 03_CONTIGS/NR_MAGs-contigs.db \
-p 06_NR-MAGs-MERGED/NR_MAGs/PROFILE.db \
-C NR_MAGs \
-o 07_NR-MAGs-SUMMARY
7- Compute CheckM taxonomy for the AOA MAGs
# From the summary I generated I will extract the fasta file for each MAG separately to run CheckM on them
# go through each MAG, in each SUMMARY directory, and store a copy of the FASTA file
# with proper deflines in the REDUNDANT-MAGs directory:
awk '{print $2}' NR_MAGs_COLLECTION.txt | sort | uniq > NR_arc_MAGs_list.txt
for BIN in `cat NR_arc_MAGs_list.txt`
do
anvi-script-reformat-fasta 07_NR-MAGs-SUMMARY/bin_by_bin/$BIN/$BIN-contigs.fa \
--simplify-names --prefix $BIN \
-o 02_FASTA/$BIN.fa >> 00_LOGS/Reformat_fasta_archaea.log
done
conda activate checkm
checkm tree 02_FASTA -x .fa -t 30 CHECKM/NR-MAGs-CHECKM-TREE
checkm tree_qa CHECKM/NR-MAGs-CHECKM-TREE -f CHECKM/NR-MAGs-CHECKM-TREE.txt
checkm lineage_set CHECKM/NR-MAGs-CHECKM-TREE CHECKM/NR-MAGs-marker-file.txt
checkm analyze CHECKM/NR-MAGs-marker-file.txt 02_FASTA -x .fa CHECKM/NR-MAGs-CHECKM-TREE -t 30
checkm qa CHECKM/NR-MAGs-marker-file.txt CHECKM/NR-MAGs-CHECKM-TREE > CHECKM/table_qa.txt
# visualize the non-redundant AOA bin distribution
ssh -L 8080:localhost:8080 trouche@penduick
anvi-interactive -p 06_NR-MAGs-MERGED/NR_MAGs/PROFILE.db \
-c 03_CONTIGS/NR_MAGs-contigs.db -C NR_MAGs --server-only -P 8080
8- Create single MAGs profiles for vizualisation and further analysis
anvi-split -c 03_CONTIGS/NR_MAGs-contigs.db \
-p 06_NR-MAGs-MERGED/NR_MAGs/PROFILE.db \
-C NR_MAGs \
-o 09_SPLIT_NR_Arc_MAGs
9- Run variability profiling for all MAGs
Conditions: - all genes - 10X coverage in sample (otherwise everything is removed) - 10X coverage of genes - 10% divergence
Targets: All bins from the 4 main clades with more than 1 sample with 10X coverage (This removes MAG A7S_Bin_00141, so 42 MAGs)
# Create sample files with samples for which MAG has 10X coverage
cd NON_REDUNDANT_BINS
mkdir -p 10_VAR_PROFILES/1_sample_files
for MAG in `cat Nitroso_MAGs_main_clades.txt` # this file can be found in the config archive
do
# look at line for specific MAG, then find columns for which coverage is over 10 and write to temp file
grep $MAG 07_NR-MAGs-SUMMARY/bins_across_samples/mean_coverage.txt \
| awk '{for(i=2;i<=NF;i++){ if($i>=10){print i} } }' > tmp_colnb.txt
# read temp file and extract column sample names from input for samples > 10X coverage
while read line;
do
grep bins 07_NR-MAGs-SUMMARY/bins_across_samples/mean_coverage.txt | awk -v fld=$line '{print $fld}';
done < tmp_colnb.txt > 10_VAR_PROFILES/1_sample_files/${MAG}_samples_over10.txt
done
# generate variability profile for nucleotides: 10% divergence from ref, 10X coverage of gene
cd NON_REDUNDANT_BINS
for MAG in `cat Nitroso_MAGs_main_clades.txt`
do
# var profile, 10%
anvi-gen-variability-profile -c 03_CONTIGS/NR_MAGs-contigs.db \
-p 06_NR-MAGs-MERGED/NR_MAGs/PROFILE.db -C NR_MAGs -b $MAG \
--engine NT --min-coverage-in-each-sample 10 --quince-mode \
--samples-of-interest 10_VAR_PROFILES/1_sample_files/${MAG}_samples_over10.txt \
--min-departure-from-consensus 0.1 \
-o 10_VAR_PROFILES/2_SNV_files/SNVs_10dep_10cov_${MAG}.txt
# Here we add a further filter: detection over 0.7 in the considered samples
# input files can be found in the config archive
anvi-gen-fixation-index-matrix --variability-profile 10_VAR_PROFILES/2_SNV_files/SNVs_10dep_10cov_${MAG}.txt \
--output-file 10_VAR_PROFILES/3_FST_SNV/FST_${MAG}.txt \
--samples-of-interest 10_VAR_PROFILES/1_sample_files/${MAG}_samples_over10_over0.7.txt
anvi-matrix-to-newick 10_VAR_PROFILES/3_FST_SNV/FST_${MAG}.txt \
--output-file 10_VAR_PROFILES/3_FST_SNV/FST_${MAG}.newick
done